Autoinitialize
1 - Averaging Compounds Finite Difference Approximation
The first step is to obtain the derivative approximation for every compound in each set, considering the first and last datapoint:
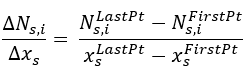
|
|---|
There, Ns,i refers to the experimental total moles values for compound i in set s. The x variable is the discretization variable: Time for Batch, Catalyst Mass or Volume for PFR.
For CSTR, the equation used is:
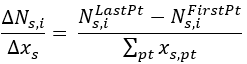
|
|---|
Where the denominator is the sum over all datapoints of Catalyst Mass or Volume values.
Then, the finite difference values are averaged among all sets:
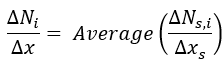
|
|---|
Finally the averaged difference values are sorted by magnitude, from the largest negative values (corresponding to reactants) to the largest positive values (products).
2 - Identifying Key Compound and Reactions
Here we identify the compound that reacts the most and mark it as the Key compound, and the key reactions where that key compound is a reactant.
That key compound will be the one with the highest negative average finite difference, that also fulfills these requirements:
- Being a reactant in at least one reaction that is estimated and have open preexponential bounds.
This criterion will rule out inerts, for which there are experimental values defined for the first datapoint but may not have values entered on the following points.
- Of the reactions considered above, any co-reactants with the key compound must have at least one set with non-zero initial value. If it turns out that all co-reactants have initial values of zero, this key compound is discarded and the next one from the sorted list is chosen and the test is repeated. All reactions with the key compound that meet the condition above on the co-reactants are considered key reactions.
- If a candidate key compound has zero measured value in the last datapoint in more than 90% of the experimental sets, then we avoid choosing that compound as key and move on to the next one in the sorted list. However, if the remaining candidates have the same issue, then the original key compound with highest finite difference is chosen as the key.
3 - Intermediate Variables
In order to get the approximation for the kinetic parameter values, we must get the compound concentration values.
Average volume (for Batch) or flow (for PFR/CSTR) are obtained based on the experimental values for first and last datapoints. In models where reaction phase volume is calculated through compound densities, or reaction phase gas flow by pressure fixed, those calculations are applied based on the total moles experimental values (initial point) to get an approximation for volume or flow.
Average Concentration ( or Partial Pressure) among all sets is then obtained.
4 - Estimating Kinetic Parameters
For all estimated reactions, we assume an activation energy of 40 kJ/mol, provided that 40 kJ/mol is within the activation energy bound range. If not, the closest value of that range to the 40 value is used as initial energy.
Then, the key component consumption is assumed to be caused only by the forward direction of the key reaction. In case there are more than one key reaction, each one is assumed to consume same amount of the key component. For example, with 2 key reactions, each key reaction will be assigned half of the consumption. The effect of the key compound consumption (or formation) in other reactions is ignored.
For example for Batch with molarity concentration units and only one key reaction:
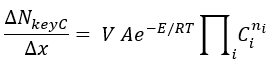
|
|---|
The total consumption NkeyC for the key component is assigned to the only key reaction for this example. V represents the batch average volume, A and E Preexponential and Activation Energy for the key reaction, and Ci are the average concentration for the compound orders for that reaction.
Thus the preexponential value (A) is obtained from there for the key reaction. For the non-key reactions, their rate value (left hand side of above equation) is assumed to be one fifth of the key compound value. Preexponentials are calculated for these non-key reactions by using their corresponding rate equations. All reverse reactions are by definition non-key, so they will be treated as mentioned above. Note that the values of pre-exponentials (A) may be outside the bounds entered by the user.
The normal solver run then proceeds from here and will honor all bounds entered by the user. In addition, the overall rate constant is bounded between 1/1000 and 1000 of the value resulting from the initialization. This is done to ensure that the kinetics do not change dramatically in the first run. After a successful initialization run, it is important to import these values using the "Estimation->Initialize from Results->Initialize All" action. This action will automatically turn OFF the autoinitialize feature. A new run can be launched from here to do a full estimation without the limiting bounds on the rate constant.
5 - Other Considerations
LHHW Sites are not taken into account. Even if your project has sites defined, they are internally disabled so the run will not report any site parameters. In Detailed Catalyst models where there are surface species, each species is assumed to have equal site coverage for the auto-initialization. For example, if we have free catalyst and 2 catalyst adsorbate complexes, then each site fraction is set to be 1/3
Top of Topic
See Also: